インシリコ創薬におけるタンパク質構造予測
この記事は約15分で読めます。
立体構造決定の歴史と各手法の比較
タンパク質の立体構造(3D空間構造)は生理学的な機能と密接に関連しています[1]。 この構造を解明することは、生物学的プロセスの理解やライフサイエンス分野の課題解決に不可欠です。 タンパク質の構造決定には、クライオ電子顕微鏡法、核磁気共鳴分光法、X線結晶構造解析などの実験技術がゴールドスタンダードとされてきました。 しかし、これらの実験手法はプロセスが複雑で、多大な時間と費用を要します。また、天然状態の構造を捉える事が難しいという課題もあります。 一方で、ゲノム解析により特定されたアミノ酸配列の数は、実験的に解明された立体構造の数をはるかに上回るスピードで増え続けました。 その結果、蓄積されたタンパク質データには大きな溝(データギャップ)が生じました[2,3]。 このデータギャップを埋めるべく急速に発展した手法が タンパク質構造予測、特に一次元アミノ酸配列から三次元立体構造を予測する分野 です。 予測技術は1970年代の手作業による最適化から始まり、1990年代の計算設計を経て、現在はAI駆動型の手法へと劇的な進化を遂げています。
| 立体構造決定手法 | 特徴や利点 | 適用限界や欠点 | 適用対象 |
|---|---|---|---|
| クライオ電子顕微鏡 (Cryo-EM) |
・結晶化が不要で生体に近い状態で観察可能 ・巨大な複合体や膜タンパク質に強い |
・分子量が小さいもの(約100-150kDa以下)は困難 ・装置が非常に高価で、高度な解析技術が必要 |
巨大タンパク質、ウイルス、膜タンパク質 |
| X線結晶構造解析 (X-ray) |
・極めて高い分解能(原子レベル)が得られる ・技術が成熟しておりスループットが高い |
・高品質な結晶が必須 (最大の難関) ・結晶中の「静止」した状態しか見られない |
あらゆるサイズの結晶化可能な分子 |
| 核磁気共鳴分光法 (NMR) |
・溶液中(自然な環境)での構造・動態がわかる ・タンパク質の「動き」の解析に最適 |
・解析できる分子量に上限がある(通常50kDaまで) ・高濃度の試料と安定同位体標識が必要 |
小分子、中分子、柔軟なタンパク質 |
| インシリコ構造予測 (HM, MD, AI) |
・実験不要でコンピュータ上で迅速に予測可能 ・未知の構造の足がかりになる |
・あくまで「予測」であり、信頼性の検証には実験が必要 ・新規性の高い構造の予測は難しい場合がある |
既知の構造に似たタンパク質 |
分子シミュレーションと併用する利点
昨今のバイオ医薬品の創薬研究では、特異性と安全性の向上、新規治療法クラスの確立、複雑な疾患メカニズムの解明、の各需要が高まっています[4,5]。 これらの需要に対応するためには、治療薬の作用機序への深い理解が必要です。 標的分子と薬剤分子の非共有結合相互作用、結合安定性、立体構造変化、溶媒和などの解析には 分子シミュレーション が有用です[6]。 代表的手法の1つである分子動力学シミュレーションは、計算手法の進歩と計算資源の拡充によって、実験と同等の精度での解析が可能になりました[7]。 また、洗練された計算手法である分子ドッキングシミュレーションでは、タンパク質複合体の迅速な静的解析が可能です[8,9]。 そして、これらの分子シミュレーションはタンパク質構造予測と組み合わさる事で、分子データが有効に活用されます。
BIOVIA Discovery Studio は、タンパク質の構造予測、分子動力学または分子ドッキングのシミュレーション、結果の解析をシームレスに利用できるツールセット を提供します。
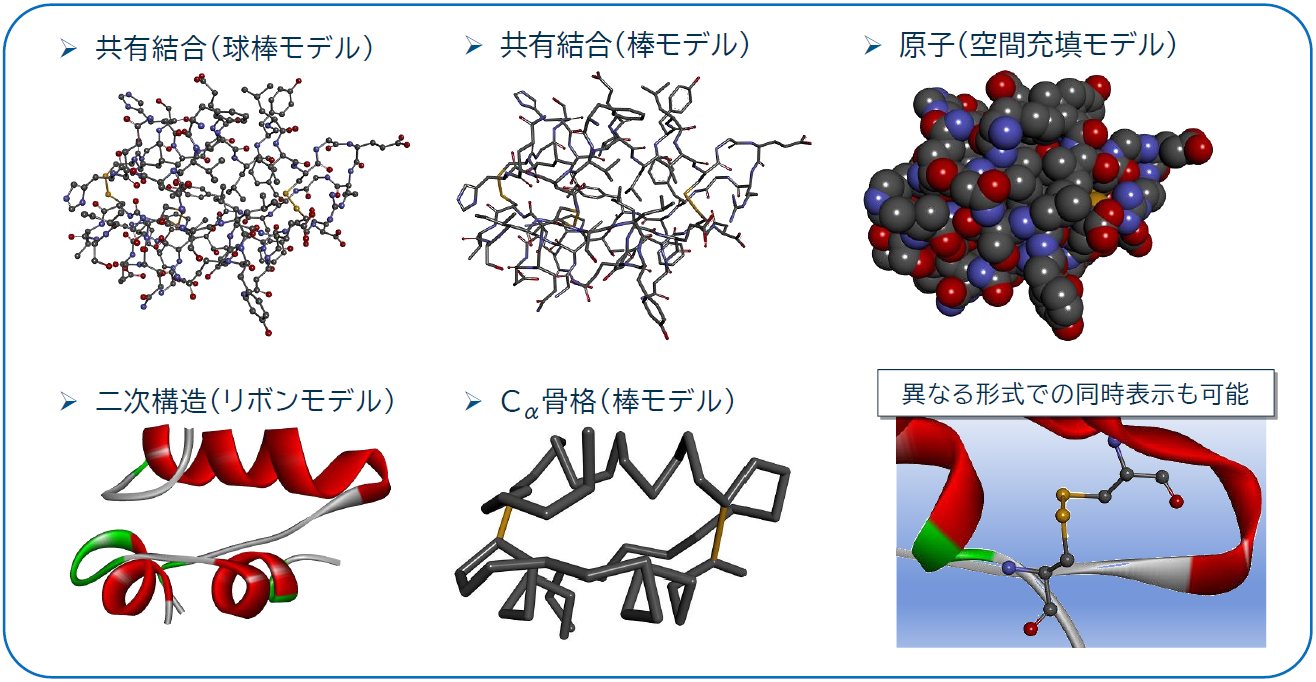
ホモロジーモデリングによるタンパク質構造予測
手法の概要
ホモロジーモデリングは、タンパク質のアミノ酸配列から立体構造を決定する代表的なインシリコ予測技術の1つです。 特に構造ベース創薬においては、標的タンパク質の立体構造を準備するための標準的な手法として広く活用されています[10]。
ホモロジーモデリングでは、立体構造が既知の類似タンパク質を鋳型(テンプレート)として、クエリとなるタンパク質の構造を予測します[11]。 その予測精度は、クエリ配列とテンプレート配列が持つ相同性(ホモロジー)の高さに大きく左右されます[12]。 また、クエリ配列をテンプレートの空間位置に正しくマッピングするための、配列の位置合わせ(アラインメント)の品質も予測精度に大きく影響します[13]。
ホモロジーモデルの典型的な構築手順は、次の5つのステップで構成されます。
- 配列準備
構造を予測したいタンパク質のアミノ酸配列を用意します。 - テンプレート同定
配列ホモロジーを検索して最適なテンプレートを選択します。 - アラインメント
クエリ配列とテンプレートの対応関係を決定します。 - モデル構築
ホモロジー法を用いて3Dモデルを複数生成します。 - モデル評価
スコアリングにより3Dモデルの妥当性を評価します。
3Dモデルが得られた後は、通常は分子動力学法などで構造の最適化を図ります。 また、ループ領域は配列の多様性が高い(すなわち、ホモロジーが低い)ために、一般的には予測精度が低下しやすい箇所です。 そのような領域に対しては、部分構造の再構築と最適化が実施されることもあります[14]。
配列ホモロジー検索とテンプレート同定
ホモロジーモデリングで重要なステップの1つに、配列ホモロジーを持つテンプレートの同定作業があります。 BIOVIA Discovery Studio は、単一または複数のタンパク質テンプレートを使用してモデリングします。 既知のタンパク質に対する配列ホモロジー検索は BLAST プログラムが一般的に用いられます[15][16]。 BLAST は、PDB登録タンパク質に関する配列データベースに対して検索を行います。
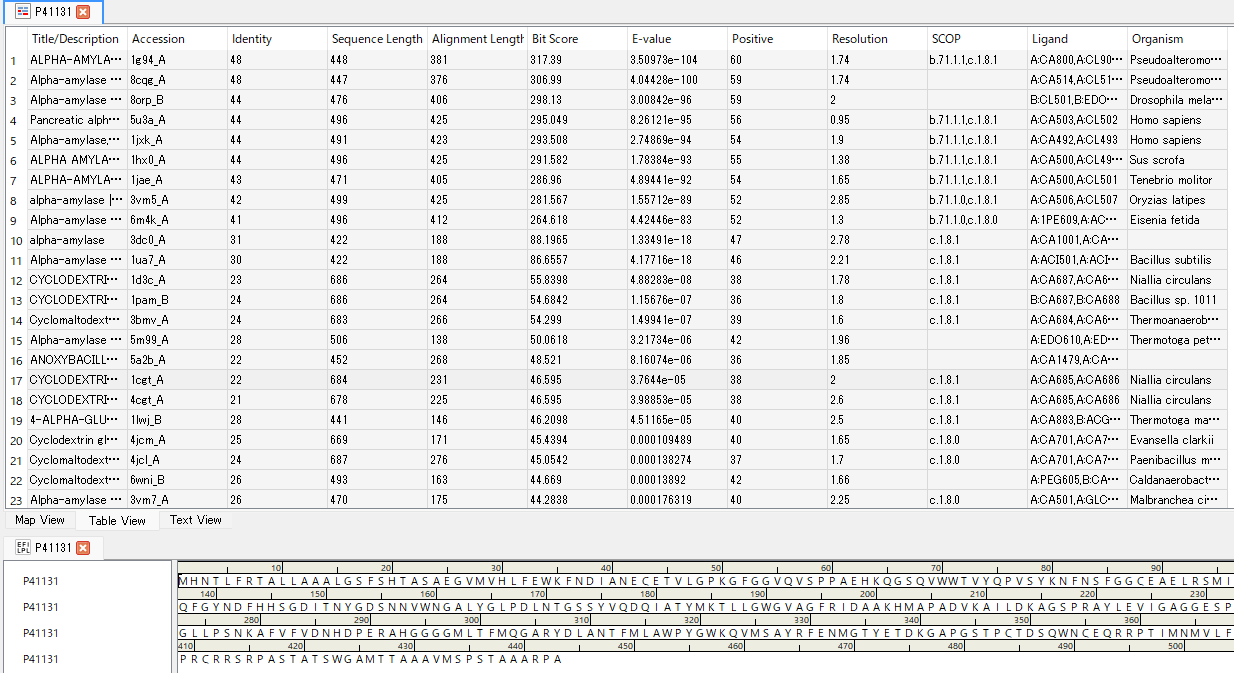
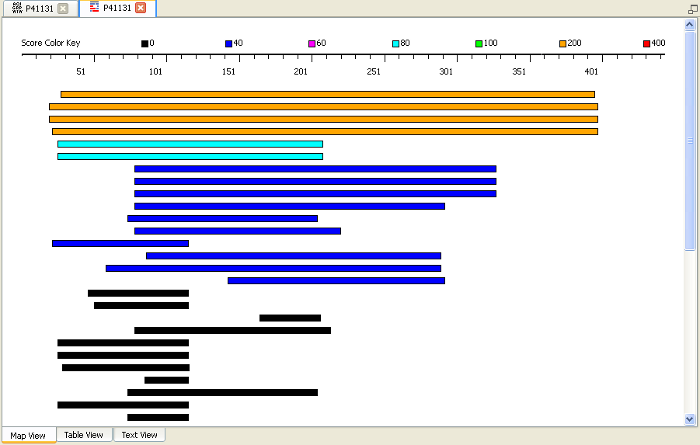
タンパク質が持つ潜在的な標的としての特徴を発見する際には、配列解析から作業を始めることがよくあります[16,17]。 同一あるいは類似の機能を持つタンパク質同士は、殆どの場合において高い配列ホモロジーを共有するため、 ホモロジー検索によって「特定の機能に関与している可能性が高い配列」を効率よく特定できます[18]。 多重配列アラインメント(MSA)などの比較手法を用いると、保存性の高い領域(進化的に変化していない重要な部位)が浮き彫りになります。 これにより、特定の配列領域が持つ機能的な重要性について、より具体的かつ高精度な予測が可能となります[19]。
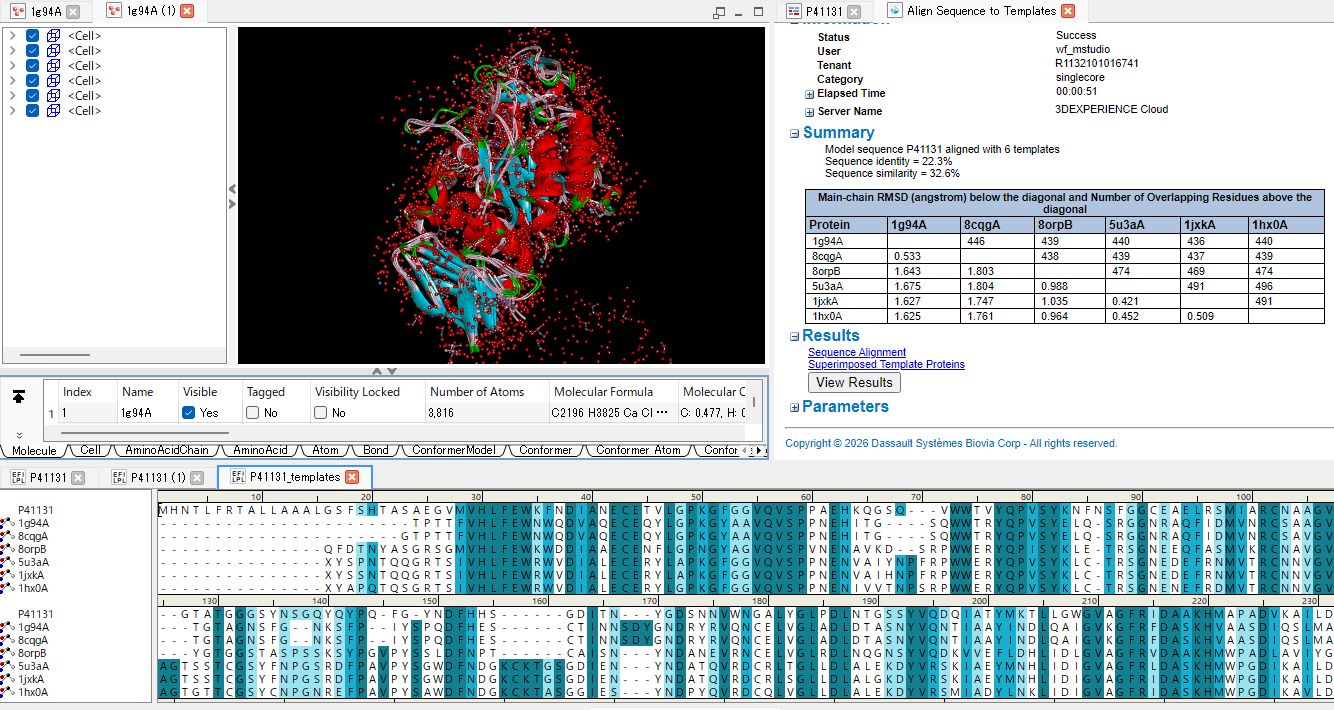
クエリ配列とテンプレートのアラインメント
クエリ配列とテンプレートのアラインメントは、最終的なホモロジーモデルの品質に直結するため非常に重要な作業です[20]。 なぜなら、ホモロジー認識から3Dモデル構築に至るまで、全ての計算はこのアラインメントの結果を基準に行われるためです。 BIOVIA Discovery Studio では、複数のテンプレートを同時に利用する場合や、高いホモロジーを持つテンプレートが見つからない場合、 といったシナリオにも対応できるように、多彩なアラインメント手法が用意されています。 状況に応じて、次のような手法を選択・組み合わせることが可能です。
- ペアワイズ配列アラインメント[21]
クエリと高いホモロジーを持つ単一のテンプレートを使用する場合に最適です。 - プログレッシブ法による多重配列アラインメント[22]
類似性の高い配列から順番に、段階的にアラインメントしていく手法です。 - 構造類似性を用いた配列アラインメント[23]
配列ホモロジーが低い領域については、立体構造の類似性からアラインメントする方法も提案されます。 - 配列プロファイルを用いた多重配列アラインメント[24]
配列群の特徴情報(プロファイル)を利用することで、より高精度な多重整列が可能です。
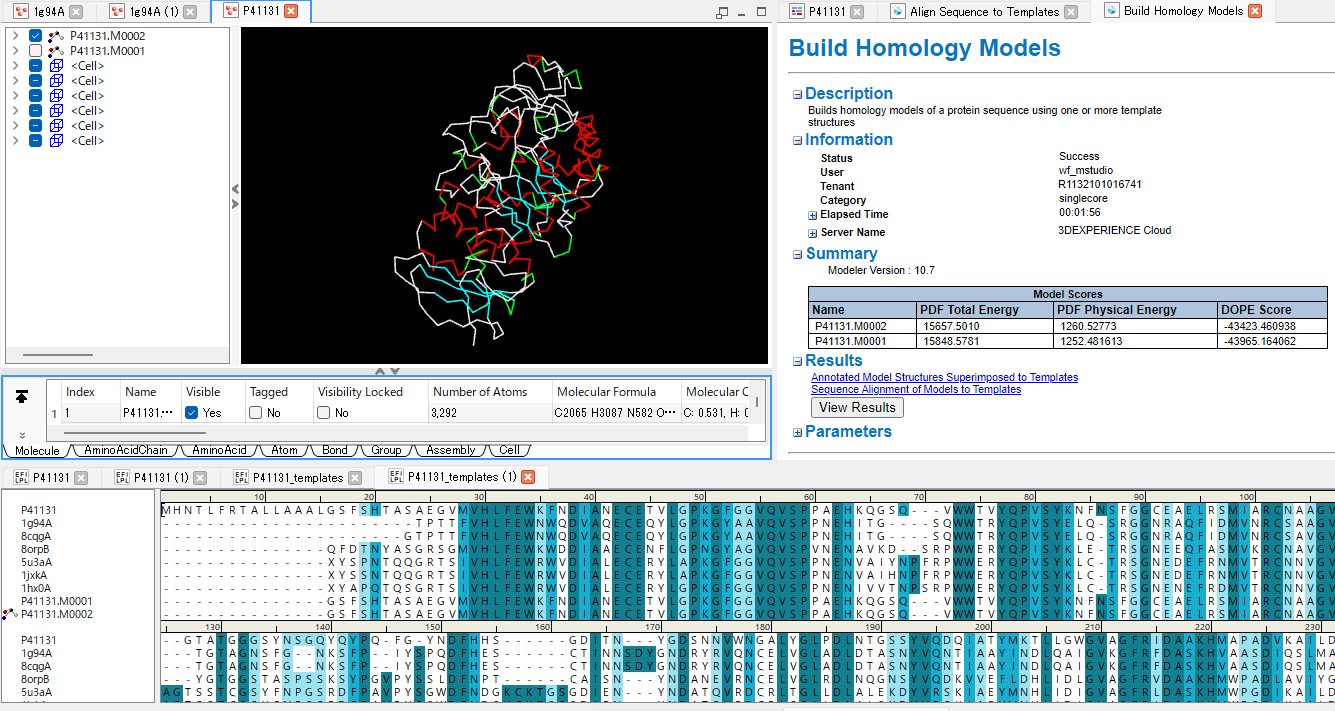
ホモロジー法による3Dモデルの構築と検証
BIOVIA Discovery Studio の基本機能として組み込まれている MODELLER プログラムは、 配列のホモロジーとテンプレート構造の特徴から空間的制約を導出し、 これを基に共役勾配法およびシミュレーテッドアニーリング最適化手順を用いて3Dモデルを複数作成します[11,25]。 作成された立体構造に対しては、DOPEスコアや配列整合性スコアなどの多角的な評価で検証可能です[26,27]。
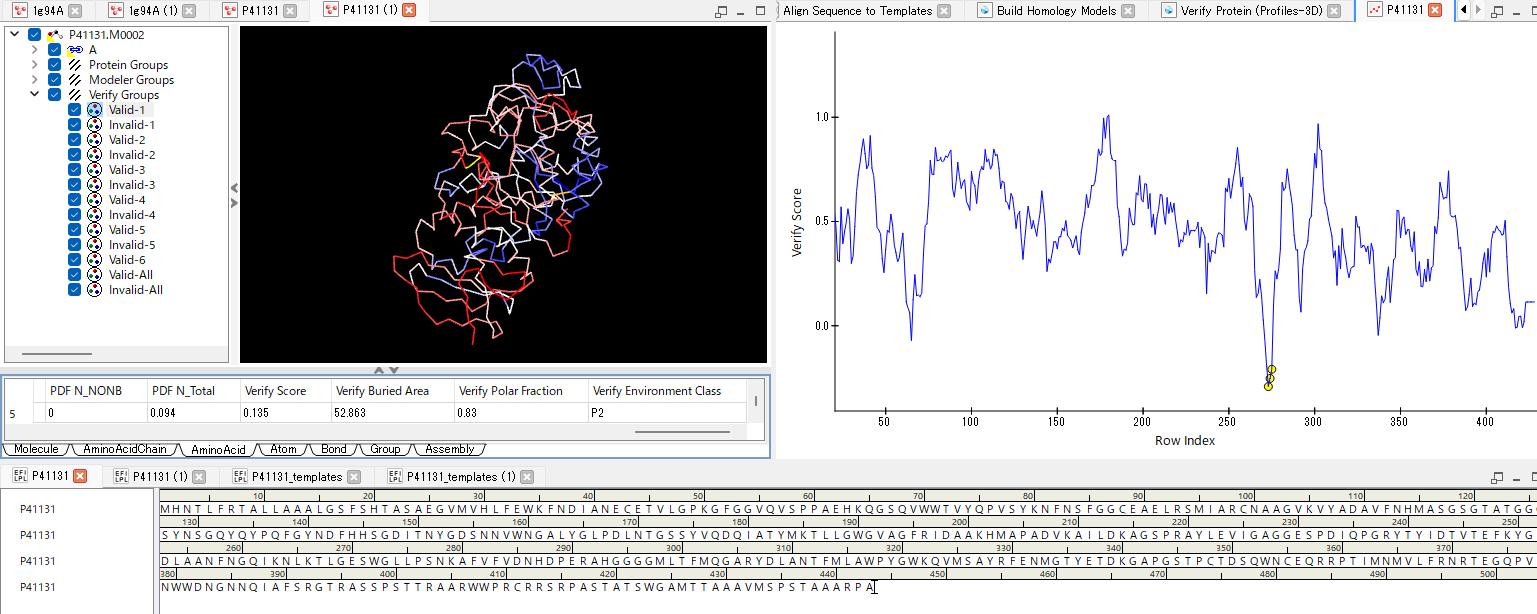
AIアシストタンパク質モデリング
タンパク質構造予測の最先端手法に大きな進展がありました。 Google DeepMind の AlphaFold は、深層学習の技術を使用して作成されたタンパク質構造予測のAIプログラムであり、 立体構造予測の国際コンテスト CASP13 (2018年) で優勝しました[28,29]。 更にその改良版である AlphaFold2 は、CASP14 (2020年) において競合手法を上回る圧倒的な性能を発揮して優勝し、 これによりAIベース手法の優位性が実証されました[30-34]。
BIOVIA Discovery Studio には、最先端の生成AIプログラムが複数組み込まれています。 指定されたアミノ酸配列から立体構造を予測するツールには OpenFold と AlphaFold2 の2つが採用されています[33,35]。 多量体配列の構造予測には AlphaFold2 が使用され、単量体配列の構造予測にはGPU処理に最適化された OpenFold が使用されます。 これらは内部処理で自動的に切り替わります。

また、2025年には3種類の生成AIを組み合わせた「タンパク質結合剤のデノボ設計」ツールが実装されました。 これは、標的タンパク質に結合するタンパク質の骨格を RF Diffusion で多数生成し、 それらの配列を ProteinMPNN(あるいは、その拡張・派生である LigandMPNN または SolubleMPNN)で設計し、 AlphaFold2 で立体構造の再構築・最小化・検証を行います[36,37]。 スコアリングとフィルタリングには RMSD, pLDDT, PAE 等を組み合わせて設定可能です。 これらの多段階AIワークフローはパイプライン処理で連続実行することも可能ですし、段階別に個別実行することも可能です。

さらに2026年には、OpenFold3 を用いた「タンパク質とリガンドの構造(コフォールディング)を予測する」 ツールも新規実装される予定です。今後の BIOVIA Discovery Studio の進化にも是非ご期待ください。
関連記事

参考文献
C. B. Anfinsen (1973), Science, 181(4096): 223-230, DOI:10.1126/science.181.4096.223
KB statistics, UniProt, URL:uniprot.org/uniprotkb/statistics
PDB Statistics, RCSB PDB, URL:rcsb.org/stats
G. Walsh (2018), Nat. Biotechnol., 36(12): 1136-1141, DOI:10.1038/nbt.4305
A. Mullard (2021), Nat. Rev. Drug Discov., 20(2): 77-81, DOI:10.1038/d41573-021-00002-0
M. Karplus and J. A. McCammon (2002), Nat. Struct. Biol., 9(9): 646-652, DOI:10.1038/nsb0902-646
S. A. Hollingsworth and R. O. Dror (2018), Neuron, 99(6): 1129-1143, DOI:10.1016/j.neuron.2018.08.011
G. Klebe (2000), "Virtual Screening: An Alternative or Complement to High Throughput Screening?" 2002nd, Springer, ISBN:978-0-7923-6633-1
D. B. Kitchen, et al. (2004), Nat. Rev. Drug Discov., 3(11): 935-949. DOI:10.1038/nrd1549
C. N. Cavasotto and S. S. Phatak (2009), Drug Discov. Today, 14(13-14): 676-83, DOI:10.1016/j.drudis.2009.04.006
A. Sali and T. L. Blundell (1993), J. Mol. Biol., 234(3): 779-815, DOI:10.1006/jmbi.1993.1626
C. Chothia and A. W. Lesk (1986), EMBO J., 5(4): 823-826, DOI:10.1002/j.1460-2075.1986.tb04288.x
B. Rost (1999), Protein Eng., 12(2): 85-94, DOI:10.1093/protein/12.2.85
A. Fiser, R. K. G. Do, and A. Sali (2000), Protein Sci., 9(9): 1753-1773, DOI:10.1110/ps.9.9.1753
S. F. Altschul, et al. (1990), J. Mol. Biol., 215(3): 403-410., DOI:10.1016/S0022-2836(05)80360-2
S. F. Altschul, et al. (1997), Nucleic Acids Res., 25(17): 3389-3402, DOI:10.1093/nar/25.17.3389
I. Gashaw, et al. (2011), Drug Discov. Today, 16(23-24): 1037-1043, DOI:10.1016/j.drudis.2011.12.008
S. R. Eddy (1998), Bioinformatics, 14(9): 755-763, DOI:10.1093/bioinformatics/14.9.755
J. D. Thompson, et al. (1994), Nucleic Acids Res., 22(22): 4673-4680, DOI:10.1093/nar/22.22.4673
R. Sanchez and A. Sali (1997), Curr. Opin. Struct. Biol., 7(2): 206-214, DOI:10.1016/s0959-440x(97)80027-9
S. B. Needleman and C. D. Wunsch (1970), J. Mol. Biol., 48(3): 443-453, DOI:10.1016/0022-2836(70)90057-4
D. F. Feng and R. F. Doolittle (1987), J. Mol. Evol., 25(4): 351-360, DOI:10.1007/bf02603120
I. N. Shindyalov and P. E. Bourne (1998), Protein Eng., 11(9): 739-747, DOI:10.1093/protein/11.9.739
M. Gribskov, A. D. McLachlan, and D. Eisenberg (1987), Proc. Natl. Acad. Sci. USA, 84(13): 4355-4358, DOI:10.1073/pnas.84.13.4355
A. Sali and J. P. Overington (1994), Protein Sci., 3(9): 1582-1596, DOI:10.1002/pro.5560030923
M. Y. Shen and A. Sali (2006), Protein Sci., 15(11): 2507-2524, DOI:10.1110/ps.062416606
N. Eswar, et al. (2006), Current Protocols in Bioinformatics, 15(1): 5-6, DOI:10.1002/0471250953.bi0506s15
A. Kryshtafovych, et al. (2019), Proteins, 87(12): 1011-1020, DOI:10.1002/prot.25823
A. W. Senior, et al. (2020), Nature, 577(7792): 706-710, DOI:10.1038/s41586-019-1923-7
Protein Structure Prediction Center (2020), CASP14, URL:predictioncenter.org/casp14/
Science blog (2020), "AlphaFold: a solution to a 50-year-old grand challenge in biology.", Google DeepMind, URL:deepmind.google/blog/
J. Pereira, et al. (2021), Proteins, 89(12): 1687-1699, DOI:10.1002/prot.26171
J. Jumper, et al. (2021), Nature, 596(7873): 583-589, DOI:10.1038/s41586-021-03819-2
K. Tunyasuvunakool, et al. (2021), Nature, 597(7877): 427-431, DOI:10.1038/s41586-021-03828-1
G. Ahdritz, et al. (2024), Nat. Methods, 21(5): 770-778, DOI:10.1038/s41592-024-02272-z
J. L. Watson, et al. (2023), Nature, 620(7976): 1089-1100, DOI:10.1038/s41586-023-06415-8
J. Dauparas, et al. (2022), Science, 378(6615): 49-56, DOI:10.1126/science.add2187
記事制作者
- 執筆:
- ララサティ・マルタ (株式会社ウェーブフロント 連成問題研究部)
- 佐橋 一裕 (株式会社ウェーブフロント 連成問題研究部)
- 編集:
- 佐橋 一裕 (株式会社ウェーブフロント 連成問題研究部)